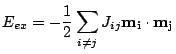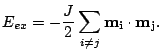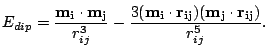Next: 2.2.3 Micromagnetics: classical approach Up: 2.2 Linking different spatial Previous: 2.2.1 Ab-initio calculation of Contents
In this approach every physical magnitude has its value in the atom
sites and atomic-level information, such as interatomic distance or
magnetic moment, is explicitly used. Although the ab-initio
simulations are also atomistic, this paragraph only refers to the
simulation based on semiclassical energy. The spin is a vector whose
components can have any value. In fact the true classical approach is strictly speaking valid for
![]() , being
, being ![]() the spin. However, in this kind of
atomistic simulations some quantum features can be added, for
example discrete values for the spin projection, a classical example for this is the Ising model that represents a spin
the spin. However, in this kind of
atomistic simulations some quantum features can be added, for
example discrete values for the spin projection, a classical example for this is the Ising model that represents a spin ![]() .
Contrary to the ab-initio models, the atomistic simulations do not
consider the electronic structure. The atomistic models have been
extensively used in other fields of the materials science as in film
growth dynamics [Rahman 04], solid-liquid systems [Hoyt 02],
etc.
.
Contrary to the ab-initio models, the atomistic simulations do not
consider the electronic structure. The atomistic models have been
extensively used in other fields of the materials science as in film
growth dynamics [Rahman 04], solid-liquid systems [Hoyt 02],
etc.
The atomistic sites have a localized magnetic moment ![]() , whose
value is obtained from experiments or ab-initio calculations. The
natural unit is the Bohr magneton
, whose
value is obtained from experiments or ab-initio calculations. The
natural unit is the Bohr magneton
![]() and the moment constitutes usually few magnetons. Instead of the vector
and the moment constitutes usually few magnetons. Instead of the vector
![]()
![]() , it is more convenient to work in
reduced magnetic moment
, it is more convenient to work in
reduced magnetic moment
![]()
![]()
![]() .
.
From the magnetic point of view, the main energy contributions are the Zeeman energy, the magnetic dipole-dipole energy, the exchange energy and the magnetic anisotropy energy.
The anisotropy reflects the existence of preferred spatial directions in
which the magnetization will align. There are different sources of
anisotropy, but their contribution to the energy can always be
written in similar functional dependence on
![]() . The
principal source is the magnetocrystalline anisotropy that has its
origin in the spin-orbit (LS) coupling and the ionic crystal field.
Due to this fact, the magnetocrystalline anisotropy has the same
symmetry as the lattice of the material. For uniaxial materials the
anisotropy energy is an expansion of even powers of
. The
principal source is the magnetocrystalline anisotropy that has its
origin in the spin-orbit (LS) coupling and the ionic crystal field.
Due to this fact, the magnetocrystalline anisotropy has the same
symmetry as the lattice of the material. For uniaxial materials the
anisotropy energy is an expansion of even powers of ![]() that usually
reduces to its first term:
that usually
reduces to its first term:
The Zeeman energy is the energy of a magnetic dipole in a magnetic field:
The exchange interaction is an electronic interaction whose origins are the Pauli exclusion principle and the Coulomb repulsion. The exchange interaction requires the integration of the electronic wave function. A more convenient evaluation of the exchange generally involves calculating the exchange explicitly using the Heisenberg form:
The interaction energy between two moments in the material is the classical dipole-dipole energy:
2008-04-04